心血管疾病是人类死亡的首要原因,而心力衰竭(HF, heart failure)是所有是心脏疾病发展的终末阶段。其中高血压和心室血液流出受阻造成的心脏压力超负荷是导致心脏衰竭主要原因之一。
在长期压力负荷下,心脏重塑缓慢发生,除心肌纤维增长、室间隔和室壁厚度增加、纤维化程度增加外,炎症反应也贯穿其中,最终导致心脏搏出功能降低。压力超负荷的HF后期心脏具有局部T细胞的活化的免疫学特征,但其更早期的诱发因素并不清楚。
巨噬细胞是心脏中起源最早、数量最多的免疫细胞,在维持健康与病理状态下心脏的稳态平衡具有关键的作用。健康小鼠心脏中巨噬细胞占非心肌细胞的6-8%,其中80%为胚胎起源的CCR2-巨噬细胞,20%为成年造血干细胞起源的CCR2+巨噬细胞。在缺血性HF发生的过程中,外周血和脾脏来源的CCR2+的单核/巨噬细胞受到心脏局部CCL2的趋化到达损伤局部,大量替代原有的CCR2-巨噬细胞。这些细胞一方面通过吞噬功能清理组织局部碎片,一方面放大炎症反应加剧损伤。而在损伤缓解后的修复期,CCR2+巨噬细胞趋化减少、消耗增加,逐步又被不断原位增殖的CCR2-巨噬细取代,最终达成炎症消退与损伤修复。因此,CCR2-/CCR2+巨噬细胞的更替在缺血性HF的发生过程中具有重要的作用,但其在于压力超负荷HF发生中的作用仍少有报道。
阿拉巴马大学伯明翰分校的Sumanth D. Prabhu等2018年在《美国心脏病学会杂志:基础转化科学》杂志上发表的题为“CCR2+单核衍生巨噬细胞胞促进压力超负荷介导心室重塑”(CCR2+ Monocyte-Derived Infiltrating Macrophages Are Required for Adverse Cardiac Remodeling During Pressure Overload)证明:CCR2+单核/巨噬细胞促进心脏压力超负荷诱导的心室重塑与T细胞扩增,以同时影响后期心衰的转归。
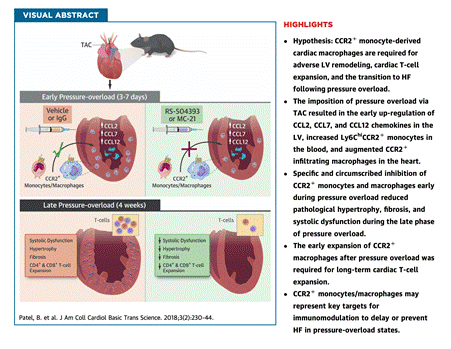
在这项研究中,作者通过对小鼠实施主动脉缩窄手术(TAC)建立压力超负荷HF模型。对TAC建模7天后心脏组织的RNA分析表明,CCL2及CCL7等趋化因子表达显著升高,通过对解离心脏的流式分析表明,TAC建模后心脏局部CCR2+单核/巨噬细胞显著升高,且CCR2+/CCR2-比例开始发生倒置。而对周血单核细胞的流式分析显示,TAC后周血中CCR2+Ly6chi的炎性单核细胞显著增加,而CCR2+Ly6clow的抑炎性细胞则有下降趋势。这提示通过阻断CCR2功能,抑制炎性单核/巨噬细胞向心脏的浸润,有望改善心脏压力超负荷的早期心脏局部的炎性环境。
应用CCR2小分子抑制剂RS504393在TAC造模后3天进行的干预研究表明,接受RS504393治疗的动物在TAC后7天(急性期),心脏局部浸润的CCR2+单核/巨噬细胞数量显著减少、心室壁增厚程度减轻、心衰标志物BNP释放减弱、心肌肥大程度减轻、CD4+与CD8+T细胞的扩增均显著减少。
而28天的长效观察结果表明,应用RS504393可显著降低TAC小鼠心脏重量增加程度、BNP释放水平及心脏纤维化水平等心力衰竭的相关指标。超声心动图分析也表明,应用RS504393治疗的TAC小鼠晚期心功能显著改善。而利用CCR2中和抗体MC21清除外周血CCR2+单核/巨噬细胞治疗TAC诱导的HF,也在体内获得了同样有效的治疗效果。
此项研究表明:CCR2+单核/巨噬细胞在早期即参与了压力超负荷导致的心脏重塑,并在HF的发生过程中具有促进作用;靶向CCR2干预外周单核/巨噬细胞的浸润有望成为压力超负荷HF的潜在治疗手段。